В современной неврологии существует огромное количество заболеваний, характер возникновений которых не поддаются рациональному объяснению со стороны специалистов. К таким болезням можно отнести группу таких недугов, как мышечная дистрофия. Всего разновидностей данной болезни девять, но обо всем по порядку…
Мышечная дистрофия - это хроническое наследственное заболевание в результате развития которого происходит поражение мышечной системы человека. Пораженные мышцы перестают нормально функционировать, истончаются в размерах, а в месте их расположения в организме постепенно нарастает жировая прослойка.
Разновидности мышечной дистрофии
В современной неврологии данный недуг классифицируется на девять различных болезней. Классификация заболевания связана с:
- локализацией мышечных нарушений;
- характеристиками болезни;
- агрессивностью развития;
- возрастом.
Так, мышечная дистрофия бывает:
- Дюшенна;
- миотоническая (болезнь Штейнерта);
- Беккера;
- Эрба Рота;
- юношеская форма дистрофии Эрба-Рота;
- плече - лопаточная лицевая форма (Ландузи-Дежерина);
- алкогольная миопатия;
- дистальная форма;
- миодистрофия Эмери -Дрейфуса.
Дистрофия Дюшенна
Наиболее известная форма прогрессирующая дистрофия Дюшенна (псевдогипертрофическая дистрофия, мерозин - негативная врожденная дистрофия). Данная разновидность недуга проявляется в детском возрасте начиная с двух до пяти лет. В первую очередь от данного недуга страдают мышцы нижних конечностей, которые несмотря на малоподвижный образ жизни у маленьких пациентов, постепенно увеличиваются в размерах. Данная особенность связана с нарастанием жировой ткани вместо мышц.

Слева здоровый ребенок, справа больной
Постепенно, по мере прогрессирования болезни она переходит в верхнюю часть и поражает мышцы верхних конечностей. Как правило, уже к двенадцатилетнему возрасту маленький пациент перестает полностью двигаться. Летальность у данного заболевания очень высока примерно 85–90% больных не доживают до 20-летнего возраста.
В группе риска находятся представители мужского пола, так как девочек данный недуг практически не затрагивает.
Болезнь Штейнерта
Врожденная дистрофия Штейнера не зря имеет название миотоническая, так как в результате прогрессирования болезни происходит слишком медленное расслабление мышц после их сокращения (такое явление называется миотонией).
Данное заболевание, в отличие от предыдущего распространено у взрослых, в возрасте от 20 до 40 лет. Имеют место быть и случаи прогрессирования болезни у детей, как правило, в младенческом возрасте, однако, это скорее исключение, чем правило.

На лицо признаки миотонии (приоткрытый рот и веки)
Гендерной зависимости болезнь не имеет и в равной степени от нее страдают как мужчины, так и женщины. Отмечается, что при недуге проявляется слабость мимических мышц лица, а также конечностей. Прогрессирование, в отличие от болезни Дюшенна происходит медленно.
Отличительная черта заболевания, возможность поражения не только мышц конечностей, но и внутренних мышц (сердечная мышца), что в свою очередь, представляет большую опасностью для жизни человека.
Болезнь Беккера
Данная форма болезни также долго прогрессирует, и пациент длительное время чувствует себя хорошо. Обострение недуга может произойти на фоне полученных травм или различных заболеваний нервной системы, которые своим течением ускорят развитие недуга.
В группе риска находятся люди низкого роста.
Болезнь Эрба-Рота и юношеская ее форма
Данное аутосомно - рецессивное заболевание развивается у пациентов после 20 лет, а юношеская форма у детей и подростков 11-13-летнего возраста. Прогрессирование болезни происходит по восходящему варианту, то есть сначала поражаются мышцы нижних конечностей, и постепенно происходит восхождение недуга к верхним конечностям.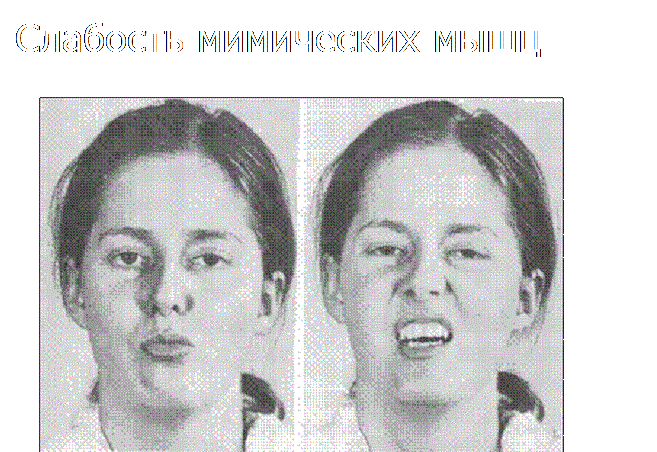
Отличительной чертой недуга является наличие выступающих лопаток, которые по мере прогрессирования выступают более явно и очевидно.
Во время ходьбы отмечается переваливание пациента, выпячивание живота и задвигание грудной клетки.
Болезнь Ландузи-Дежерина
Плече-лопаточно-лицевая форма данного заболевания является наиболее безразборным видом, так как поражает людей в возрасте от пяти до 55 лет. Для данного недуга характерно очень длительное развитие, пациент может сохранять трудоспособность до 25 лет жизни с данной болезнью.
Отличительными симптомами является поражение лицевых мышц, в результате чего у больного могут возникнуть проблемы с четкостью произношения, в связи с неполным смыканием губ. Кроме того, отмечается неполное смыкание век.
По мере развития у больного атрофируются лицевые мышцы, мышцы плеч, конечности и туловища.
Данная форма не зависит от генетической мутации.
Алкогольная миопатия
Данный вид болезни также несвязан с генными мутациями и причина его возникновения одна - злоупотребление алкоголем. У больных могут отмечаться болевые синдромы в конечностях, связанные именно с поражением мышц.
Различают острую и хроническую алкогольную миопатию.
Дистальная форма
Дистальная форма мышечной дистрофии является доброкачественным вариантом прогрессирующей дистрофии. Как правило, данное заболевание трудно дифференцировать от невральной амиатрофии Мари-Шарко. Для разделения этих двух заболеваний требуется проведение электроэнцефалограмму головы, которая и дает понимание с какой болезнью предстоит иметь дело.
К основным симптомам недуга относят атрофию мышц конечностей с их последующим исхуданием. Возможны парезы стоп, кистей рук и т. п.
Миодистрофия Эмери -Дрейфуса
Данный тип болезни первоначально, вообще, не был выделен в качестве отдельного заболевания, так как по своей симптоматике был схож с дистрофией Дюшенна. Однако, в дальнейшем в результате длительного исследования было установлено, что болезнь Эмери -Дрейфуса обладает индивидуальной симптоматикой.
Болезнь относят в разряд редких. В группе риска находятся люди до 30 лет, как правило, юный возраст. Есть свидетельства о проявлении симптомов и после 30 лет, но они единичны.
Основное отличие данного вида болезни - это проблемы с мышцами сердца, которые в итоге могут стать причиной смерти. Кардиомиопатия при данном недуге не единственное отличие. Кроме проблем с сердцем, у пациентов наблюдают стандартные для дистрофии Дюшенна признаки, но в более щадящем режиме развития.
Причины
Негативная составляющая болезней нервной системы заключается в том, что они тяжело поддаются изучению. По этой причине не до конца известно о факторах, провоцирующих развитие той или иной формы мышечной дистрофии.
Основная причина, для формирования большинства подвидов данной болезни является генная мутация, в частности гена, который отвечает за синтез белка.
К примеру, болезнь Дюшенна имеет прямое отношение к мутации половой Х хромосомы. Основной переносчик нехорошего гена выступают девушки, которые несмотря на его наличие в собственной ДНК не страдают от данной болезни.
Что касается миотонической формы, то виновник ее возникновения ген, расположенный в 19 хромосоме.
Основные симптомы
Наличие большого количество различных подвидов у данного недуга указывает на различия в симптоматике, однако у болезни есть общие симптомы, которые в себя включают:

Диагностика мышечной дистрофии
Диагностические мероприятия при подобном недуге обширны, так как существует большое количество болезней, от которых необходимо дифференцировать болезнь.
На первоначальном этапе доктор обязательно изучит анамнез больного, уточнит о симптоматике, режиме дня и т. п. Эти данные требуются для составления плана проведения последующих диагностических мер, которые могут в себя включать:

Стоит отметить, что чем позже проявилась болезнь, тем лучше для больного, так как ранние симптомы в большинстве случаев заканчиваются смертью.
Лечение
Лечение мышечной дистрофии процесс сложный и затяжной, однако, в настоящее время не создано лекарство, которое полностью исцеляет больного. Все мероприятия направлены на облегчение жизни больного и восстановления некоторых утраченных способностей.
Для затормаживания развития болезни назначают следующие препараты:
- кортикостероиды;
- витамины В1;
- аденозинтрифосфат (АТФ).
Кроме того, для замедления процесса развития используют фетальные стволовые клетки, которые замедляют процесс дистрофии.
Помимо того, в качестве профилактических мероприятий назначают:
- массаж;
- физиотерапию;
- дыхательную гимнастику.
Помимо стандартных вариантов лечения, важно постоянно руководствоваться тремя главными составляющими в процессе жизнедеятельности:
- Адекватная физическая активность.
- Своевременная психологическая поддержка.
- Соблюдение диеты.
Физическая активность
Отсутствие желания у человека бороться с недугом оказывает негативное воздействие на организм. Судите сами, пассивность, нежелание двигаться угнетает и без того пораженную мышечную систему. Мышцам необходимо давать нагрузку, так как без нагрузки дистрофичные процессы начинают происходить быстрее, тем самым прогрессируя в более быстром темпе.
Умеренная физическая активность, использование поддерживающих приспособлений будут отличным подспорьем в борьбе с недугом.
При наличии болей в мышцах отлично подойдет плавание, йога, упражнения на растяжку.
Психологическая поддержка
Психологическая поддержка окружения важна для больного человека. А если недуг такой серьезный, как этот тем более. Для кого-то будет достаточно обычной поддержки со стороны друзей и родных, а кому-то может потребоваться квалифицированная психологическая помощь.
Важно дать понять такому человеку, что он не остался один на один со своей проблемой. Он должен понимать, что ему есть к кому обратиться, есть люди, которые сопереживают и поддерживают его.
Диета
Что касается режима питания и соблюдения диеты, существует расхожее мнение, что соблюдение противовоспалительной диеты может замедлить прогрессирование недуга. Данная диета снижает воспаление, уровень глюкозы, выводит токсины из организма и питает его полезными веществами.
Суть такой диеты в следующем:
- Отказ от продуктов, содержащих «плохие» жиры и замена их на «хорошие», введение в рацион ненасыщенных жиров, которые содержатся в оливковом, льняном, кунжутном масле, авокадо.
- Применение в пищу мяса и рыбы, при производстве которых не были использованы антибиотики или гормоны.
- Полное удаление из рациона рафинированного сахара и глютена.
- Употребление в пищу следующих продуктов - китайская капуста, брокколи, сельдерей, ананас, лосось, свекла, кукумария, имбирь, куркума и других продуктов, обладающих противовоспалительными свойствами.
- Молочные продукты допустимы только на основе овечьего и козьего молока.
- Допустимо употребление травяных чаев, лимонада, кваса, морсов и натуральных соков.
Профилактика
Так как мышечную дистрофию довольно тяжело предсказать и обнаружить на раннем этапе, профилактические мероприятия сводятся к двум простым рекомендациям:
Обязательное обследование женщины на этапе планирования беременности на предмет наличия мутаций в генах
В случае если до беременности, по каким-либо причинам не было сделано генетического обследования, уже в период беременности проводится тест на определение у плода мутаций в Х хромосоме.
Прогноз и осложнения
В зависимости от вида болезни прогноз может быть разным, и тем не менее можно выделить несколько возможных осложнений, возникающих при данном недуге.
- нарушения сердечной деятельности
- искривление позвоночника
- снижение интеллектуальных способностей больного
- снижение двигательной активности
- нарушения дыхательной системы
- летальный итог
Итак, мышечная дистрофия довольно опасное и неизлечимое заболевание, поэтому будущим родителям необходимо с глубокой ответственностью относиться к планированию беременности. Берегите себя и своих будущих детей!
«Я не смогу это пережить...», - первая мысль, которая приходит в голову матери, узнавшей подробности о болезни ее ребенка. До постановки диагноза большинство даже не знает, что это за заболевание - миопатия Дюшенна (также известна как дистрофия и миодистрофия Дюшенна). И неудивительно, ведь частота встречаемости достаточно низкая: всего 1 случай на 3500 рождений мальчиков. Крайне редко эта генетическая болезнь встречается и у девочек. Но что вам до редкости заболевания, когда в карточке вашего малыша появляется запись, которую вам хотелось бы бесследно стереть?
Не упустить время
Милая неуклюжесть, сопровождающая малыша до трех лет, к пяти превращается в мышечную слабость. В 10-12 лет ребенок перестает ходить, к 15 - частично или полностью теряет способность двигаться самостоятельно. Всего 20 лет жизни - так мало, что в это невозможно поверить.
Врачи отделываются общими фразами, и родителям приходится искать информацию самостоятельно. Доктора, впервые столкнувшиеся с таким диагнозом, и сами зачастую не представляют, что делать.
Миопатия Дюшенна - заболевание, которое характеризуется мутацией в гене, отвечающем за синтез белка дистрофин в мышечных волокнах, что приводит к нарушению их строения. Носители мутаций - женщины, которые сами миопатией Дюшенна не болеют, но с 50% вероятностью передают ее своим потомкам. У мальчиков при этом развивается мышечная дистрофия, а девочки, в свою очередь, становятся носителями мутации. В некоторых случаях заболевание не является результатом передачи по наследству, а возникает спонтанно.
В России гигантский провал в данной области научных исследований. Специалистов по нейромышечным патологиям крайне мало, попасть к ним сложно. Только представьте, как много значит один год для прогрессирующего заболевания! А именно столько в среднем длится очередь в федеральное медучреждение.
Обычно сначала родители из-за трудностей при ходьбе обращаются к ортопедам, которые могут просто не знать об этом заболевании. Как показывает практика российских поликлиник, даже неврологи во многих случаях ошибаются с диагнозом. Тем более, что у 30% мальчиков миопатия сопровождается задержкой умственного развития.
Судя по частоте заболевания, в России должно быть около 4000 детей с дистрофией Дюшенна. На учете в московском детском нервно-мышечном центре - в 10 раз меньше. Где все остальные пациенты?
В этом-то и кроется самая большая проблема отечественной медицины. Невролог, наконец-то правильно поставивший диагноз, вольно или невольно убеждает родителей, что миопатия Дюшенна не лечится, быстро прогрессирует и всегда приводит к смерти. В результате - родители просто опускают руки, никуда не едут и ни к кому не обращаются, пытаясь просто выжить в поставленных условиях.
Правда ли, что ситуация настолько безвыходна? Неужели нет надежды на то, что ребенку действительно помогут? Разве неизлечимое заболевание - это повод отказаться от поисков наиболее эффективного лечения?
Учиться и работать с дистрофией Дюшенна - реально
Будем честны. Пока чуда мгновенного излечения ждать не стоит, кто бы вам что ни обещал. Однако продлить жизнь ребенка и в разы улучшить ее качество - возможно уже сейчас! Велик шанс, что за те годы, что вы ему подарите, ученые наконец-то найдут средство остановить или повернуть вспять развитие заболевания.
Центр комплексного лечения нейромышечных нарушений Детской больницы Цинциннати (Comprehensive Neuromuscular Center, Цинциннати, США) доказывает это каждый день:
Здесь регулярно проводят клинические испытания новых лекарств и методов, что позволило достичь отличных результатов в лечении мышечной миопатии Дюшенна.
На данный момент в Центре наблюдаются около 600 детей с этим заболеванием. Сюда приезжают со всего мира, чтобы получить современное лечение и эмоциональную поддержку. Медицинская команда оказывает своим пациентам и их семьям долгосрочную помощь.
Наблюдения за пациентами 13-16 лет, которые выполняли все назначения врачей Центра, подтверждают: к этому возрасту 40% из них могут самостоятельно встать с пола, 50% - пройти 10 метров без посторонней помощи. Тогда как без лечения ходьба становится абсолютно невозможной уже к 12-13 годам.
Все специалисты собраны в одном учреждении. Совместная работа неврологов, кардиологов, пульмонологов, эндокринологов, генетиков, физиотерапевтов, диетологов приводит к тому, что средняя продолжительность жизни людей с миопатией Дюшенна увеличилась на 10 лет.
Благодаря комплексному лечению и постоянному наблюдению вместо 20 лет пациенты могут жить 30 и более. Соответственно, и основные двигательные функции тоже сохраняются намного дольше.
А это значит, что дети могут успешно получить профессиональное образование и даже работать. Такие примеры среди пациентов Центра уже есть!
Эффективные методики лечения миопатии Дюшенна от Comprehensive Neuromuscular Center
Центр комплексного лечения нейромышечных нарушений работает по продуманной схеме, которая делает лечение максимально комфортным для пациента и его законных представителей (родителей или опекунов).
Преимущества лечения миодистрофии Дюшенна в Comprehensive Neuromuscular Center:
- Специализация на нейромышечных нарушениях. В Центр обращаются пациенты с миопатиями Дюшенна и Беккера, спинальной мышечной атрофией (СМА), врожденным миастеническим синдромом, наследственной атаксией Фридрейха, другими патологиями нервной и мышечной системы.
- Пациенты Центра первыми получают максимальный доступ к новым научным открытиям и достижениям, к самым последним методикам и фармацевтическим средствам. А еще - участвуют в клинических исследованиях.
- По результатам обследования лечащий врач составляет индивидуальный план в зависимости от текущего состояния ребенка и сопутствующих заболеваний. Учитывается даже образ жизни семьи в целом - все для того, чтобы предоставить наилучшую помощь.
- Управление побочными действиями. Состояние ребенка постоянно контролируется с тем, чтобы своевременно корректировать количество и вид принимаемых препаратов.
- Всесторонний амбулаторный и стационарный уход, которые выражаются в постоянной заботе. 100% ведение пациента в течение всей жизни вплоть до консультирования по вопросам качества жизни, обучения и адаптации в обществе.
Лечение миодистрофии Дюшенна будет намного результативнее, если в семье поддерживается благоприятная психологическая обстановка. Поэтому в заключение мы подготовили несколько советов, которые помогут вам сохранять спокойствие и уверенность, а ребенку - стать счастливее.
- Не скрывайте от ребенка диагноз, правдиво и понятно рассказывайте ему о заболевании.
- Помогите вашему ребенку понять, что он может и должен быть активным участником процесса лечения. Правильно питаясь и выполняя рекомендованные физические упражнения, пациент и сам положительно влияет на ход течения болезни.
- Помните, что ваш ребенок - это личность, состоящая из многих аспектов. И миопатия Дюшенна - лишь один из них, причем не самый главный.
- Обращайте внимание на то, что ребенок еще может сделать. Не зацикливайтесь на потерянных умениях.
- Решайте только актуальные проблемы. Не думайте о том, как ребенок будет чувствовать себя через месяц или через год.
- Воспитывайте ребенка так же, как здорового. Старайтесь избежать чрезмерной опеки, предоставьте ему право быть независимым в доступных для него сферах.
- Просите о помощи близких, друзей, врачей тогда, когда вам это нужно.
- По возможности ходите вместе в кино, ездите в отпуска, развлекайтесь - дарите себе и ребенку радостные моменты, наполненные позитивными эмоциями.
- Нужна подробная консультация по нейромышечным нарушениям? Отправьте запрос прямо с сайта и узнайте, как связаться со специалистами Центра комплексного лечения нейромышечных нарушений Детской больницы Цинциннати .
О таком редком и тяжелом генетическом заболевании, как прогрессирующая мышечная дистрофия Дюшенна , практически не говорят в России. Не только родители, но и многие врачи сталкиваются с ним впервые и плохо понимают, что делать с больными детьми. Между тем в настоящее время проводятся десятки международных исследований, направленных на поиски лечения.
О том, как и почему возникает это заболевание, с какими трудностями можно столкнуться при диагностике и существует ли лекарство от страшного недуга, в интервью рассказал и.о. руководителя Детского нервно-мышечного центра НИКИ педиатрии кандидат медицинских наук Дмитрий Влодавец .
Расскажите о заболевании, как оно проявляется и на что родителям стоит обратить внимание, чтобы вовремя его обнаружить?
По последним данным, с миопатией Дюшенна рождается один из 5000 мальчиков, а не один из 3000, как было принято считать раньше. Если переложить эту статистику на такой крупный город, как Москва, где за год рождаются около 100 тысяч детей, то каждый год должно рождаться 10 детей с миопатией Дюшенна.
В среднем болезнь начинает проявляться в возрасте пяти лет. Дети испытывают значительные сложности при ходьбе, быстро устают, им сложно подниматься по лестнице, а при вставании с пола они применяют миопатические приемы Говерса (вставание "лесенкой"). Еще один важный симптом - большие голени. Члены семьи поначалу радуются, думая, что ребенок растет спортсменом, однако вскоре оказывается, что это не так.
- То есть ребенок полностью здоров и вдруг резко в пять лет начинает проявляться заболевание?
Не совсем. Минимальные проявления обычно встречаются раньше, но могут просто остаться без внимания. Если же родителей подробно расспросить, как ребенок вел себя на детской площадке или во время повседневной двигательной активности, то выясняется, что он, например, так и не научился приседать, или медленно бегал, или не мог подпрыгнуть…
Только у 10 процентов пациентов встречается инфантильный тип заболевания, при котором явные клинические проявления возникают с самого рождения. В этом случае ребенок уже на первом году жизни слабый, вялый, позже других начинает ходить, позже приобретает моторные навыки.
- И как в дальнейшем развивается заболевание?
К сожалению, болезнь довольно быстро прогрессирует. У мальчиков со временем формируется гиперлордоз в поясничном отделе позвоночника (выгибание), возникает остеопороз (снижение плотности костей), контрактуры суставов (ограничение подвижности).
В среднем уже в 8-12 лет ребята теряют способность к самостоятельному передвижению. Хотя все зависит от индивидуальных особенностей. Есть мальчики, которые уже в шесть лет садятся в инвалидное кресло, а есть такие, которые ходят и в 15-16 лет.
Когда пациенты теряют возможность самостоятельно передвигаться, у них формируются новые контрактуры, в том числе коленные, тазобедренные, локтевые, межфаланговые. Еще одной проблемой становится искривление позвоночника. Ведь мальчики все равно учатся в школе, что-то пишут, читают, и, если осанка нарушена, у них может сформироваться S-образный сколиоз, который иногда требует хирургического вмешательства.
У 70 процентов пациентов к 15 годам формируется кардиомиопатия, а затем возникает прогрессирующая сердечная и дыхательная недостаточность, отчего они и погибают в возрасте 15-25 лет. Здесь также стоит отметить, что все индивидуально и некоторые пациенты доживают и до 30-40 лет.

Но почему у детей возникает эта болезнь? Что запускает генетический механизм, приводящий к столь тяжелым последствиям?
Миодистрофия Дюшенна - это генетическое заболевание, связанное с нарушением синтеза белка дистрофина, который необходим для правильной работы наших мышц. Заболевание наследуется по X-сцепленному типу наследования, так как ген, отвечающий за выработку белка дистрофина, находится на Х-хромосоме.
По молекулярным меркам это ген-великан, он состоит из 79 кусочков - экзонов. При наличии мутации в этом гене белок дистрофин в клетках мышц не синтезируется, мышечная ткань постепенно гибнет и замещается жировой и соединительной. В 60 процентах случаев мутация представляет собой делецию (потерю) или дупликацию (удвоение) одного или нескольких экзонов. В остальных случаях мы имеем дело с точечными мутациями.
- А почему миодистрофией Дюшенна болеют только мальчики?
Дело в том, что в кариотипе мужчины присутствует только одна X-хромосома, которую он получает от матери. И если он получил Х-хромосому с поврежденным геном, то дистрофин у него в организме вырабатываться не будет, соответственно, проявится миодистрофия.
У женщины всегда есть две X-хромосомы. И если на одной из них находится больной ген, то вторая здорова и производит дистрофин, поэтому заболевание не проявляется. Но не всегда мальчик получает больной ген от мамы. Примерно в 40 процентах случаев мутация возникает спонтанно в момент зачатия, при этом ни один родитель не является носителем.
Я читала, что существует не такая тяжелая форма этого заболевания, которая называется формой Беккера. В чем ее отличие на генетическом уровне?
Дело в том, что экзоны имеют разную форму. Мы можем представить ген дистрофина как пазл из 79 кусочков, вытянутых в один ряд. Если в гене отсутствуют, например, 51, 52 и 53-й экзоны, то 50-й уже не сможет соединиться с 54-м. Синтез белка начинается, доходит до 50-го экзона и останавливается.
Это называется нарушением рамки считывания и вызывает как раз миопатию Дюшенна. Но иногда рамка считывания восстанавливается самой природой. Например, в гене произошла потеря 20-го и 21-го экзонов, но форма 19-го экзона такова, что он может соединиться с 22-м. Синтез белка идет до конца, и получается немного укороченный, но вполне функциональный белок дистрофин.
Такой белок тоже работает, и заболевание протекает в более сохранной форме, которая называется формой Беккера. Она встречается реже, примерно у одного на 20 000 новорожденных мальчиков. Заболевание протекает легче, мышечная слабость возникает гораздо позднее.
Например, одному из моих пациентов с формой Беккера уже 36 лет и он живет нормальной жизнью. У него есть семья, он водит машину, работает на хорошей работе. Но у этих пациентов может быть более выражена кардиомиопатия. Бывает, что к 18 годам приходится делать пересадку сердца.
Вернемся к диагностике. Если у родителей есть подозрение на то, что у ребенка миодистрофия Дюшенна, куда им идти и какие сдавать анализы?
Да, с диагностикой в настоящее время все не так просто. В первую очередь родители обращают внимание на трудности при ходьбе и поэтому идут к ортопедам, а те, как правило, про это заболевание не знают. Так, пока ребенок попадет к неврологу, может пройти несколько лет. Да и не каждый невролог знает это заболевание!
Два года назад я стал вести базу наших пациентов. Беру у них кровь, собираю клиническую информацию: что мальчики еще могут делать, чего уже не могут, в каком возрасте садятся в инвалидное кресло. К нам приезжают со всей России и даже из сопредельных государств - Белоруссии, Украины, Киргизии, Казахстана, Таджикистана. Там врачи вообще не знают, что с такими пациентами делать, и очень их боятся.
Так вот, сейчас в моей базе 356 пациентов с миодистрофией Дюшенна и Беккера. А по некоторым расчетам, только в России должно быть около 4000 пациентов. Где они? Неизвестно. Врач-невролог по месту жительства может сказать, что ваше заболевание не лечится, ребенок скоро умрет. И родители ничего не делают. Хотя у них есть возможность обратиться в региональное отделение Минздрава - там дают бесплатное направление в Москву на обследование.
Сильно мешает диагностике задержка умственного развития, которая в той или иной степени встречается у 30 процентов пациентов. Например, полгода назад я поставил диагноз девятилетнему пациенту, у которого была очень выраженная задержка развития, и его наблюдали как пациента с аутистическим расстройством.
При миодистрофии Дюшенна креатинкиназа (КФК) в крови повышена в сотни раз! А у нас до сих пор не все врачи знают, что такое анализ КФК. Например, из Тульской области приезжает мальчик со значением КФК 25 единиц. Мы переделываем анализ, и оказывается, что у него на самом деле 25 000 единиц!
А часто КФК вообще не смотрят. В основном делают анализы АЛТ и АСТ. Это ферменты, которые в сознании врачей плотно связаны с инфекционными заболеваниями печени - гепатитами, гепатозами, циррозом печени. И, когда врач получает повышенный АЛТ и АСТ, он решает, что у ребенка гепатит. Но в данном случае АЛТ и АСТ имеют внепеченочное происхождение - они выбрасываются в кровь при разрушении мышц.
- Какие методы диагностики должны применяться в первую очередь?
Можно сделать биопсию мышечной ткани и МРТ мышц. Эти методы позволяют увидеть, что мышечная ткань замещается жировой или на месте мышц разрастаются соединительные ткани. Но так как мы имеем дело с генетическим заболеванием, для диагностики важно сделать правильный генетический анализ.
В российских лабораториях он до сих пор делается методом ПЦР, который позволяет оценить наличие только 19 экзонов. Да, это набор наиболее часто встречающихся мутаций, но не более того. Поэтому к нам приходит очень много пациентов якобы без мутаций. У них есть результат исследования, в котором написано, что мутация не обнаружена. А она у них есть, и, пока идут поиски, заболевание прогрессирует.
Существует современный тест MLPA, который позволяет оценить состояние всех 79 экзонов. Раньше мы сотрудничали с американской лабораторией в Юте, но два года назад сами стали делать его на хорошем уровне, который вполне сопоставим с зарубежными лабораториями.
- Если мутация обнаружена, что делать дальше? Можно ли помочь ребенку, существует ли поддерживающее лечение?
Во-первых, хорошо себя зарекомендовала гормональная терапия - если вовремя назначить глюкокортикостероиды, то можно добиться пролонгации самостоятельного хождения на два-три года. При регулярном применении снимаются отек и воспаление, связанные с гибелью мышечных клеток, стабилизируется мышечная мембрана, что позволяет сохранить некоторое количество клеток. Обычно назначается один из двух препаратов - преднизолон или дефлазакорт. Дефлазакорт вызывает меньше побочных действий, но пока препарат не зарегистрирован на территории РФ.

Не так давно была доказана эффективность назначения ингибиторов АПФ для профилактики дилатационной кардиомиопатии. Это те препараты, которые обычно пьют бабушки для снижения давления. Однако наши коллеги из Института миологии в Париже провели исследование, которое показало, что при раннем назначении ингибиторов АПФ к 15 годам кардиомиопатия сформировалась всего у 20-30 процентов пациентов, страдающих мышечной дистрофией Дюшенна (вместо 70 процентов, как было раньше). С этой же целью назначаются препараты, снижающие частоту сердечных сокращений.
Для профилактики остеопороза показано назначение препаратов, содержащих витамин D3 и кальций.
Обязательно нужно делать специальные растяжки ежедневно утром и вечером, а на ночь надевать тутора на голеностопные суставы. Это очень важно, однако родители не придают этому значения.
На одном приеме у меня был папа больного мальчика - военный из Архангельска.Я показал ему, как делать растяжки, все объяснил. Через год они приезжают - отличное состояние суставов, даже лучше, чем было. Проходит еще год - ребенок продолжает ходить! Оказалось, что папа ежедневно, старательно, в точности выполняет все указания. А кто-то говорит: у нас ребенок хныкал и мы перестали делать. И результат соответствующий…
Часто дети перестают ходить не из-за мышечной слабости как таковой, а из-за жутко запущенных контрактур, с которыми не работают родители или врачи - физические терапевты.
Но все же, несмотря на глюкокортикостероиды и растяжки, заболевание прогрессирует и его невозможно остановить. Или возможно? Я знаю, что в настоящее время проходят различные эксперименты, например, испытывается метод лечения под названием экзон-скиппинг. Что это такое и действительно ли он лечит мутированный ген?
Слово skip по-английски означает "прыжок". Идея заключается в том, что "перепрыгивание" определенных экзонов в гене приводит к восстановлению рамки считывания. А это значит, что в клетках начинает вырабатываться укороченный дистрофин и болезнь переходит в более сохранную форму Беккера.
Две зарубежные компании - Prosensa (Нидерланды) и Sarepta (США) - проводили клинические испытания экзон-скиппинга 51-го экзона, под которые подходили пациенты с определенными делециями - приблизительно 13 процентов Дюшеннов. Мы принимали участие в третьей фазе клинических испытаний: из 186 пациентов со всего мира восемь были нашими. Раз в неделю в течение нескольких лет мальчикам делалась подкожная инъекция.
Однако после анализа данных оказалось, что в результате исследований не было получено никакой статистически достоверной разницы между теми пациентами, которые получали лечение, и теми, которые получали плацебо. Фирма Prosensa, которая разрабатывала препарат, обанкротилась. Сейчас эти исследования и клинические испытания по экзон-скиппингу продолжает американская компания Sarepta.
Теоретически этот метод подходит только для пациентов с часто встречающимися делециями, что составляет примерно половину больных мальчиков. Разработка одного препарата для экзон-скиппинга стоит миллиарды долларов, и для пациентов с редкими делециями ее, конечно, делать не будут.
Я слышала также, что уже существует препарат "Трансларна", который очень дорого стоит и в России не продается.
Да, аталурен, или "Трансларна", подходит только для пациентов с точечными стоп-мутациями. Он способен отыскивать неправильно возникший стоп-сигнал и прочитывать ген сквозь него. Препарат действительно очень дорогой: курс лечения на ребенка весом 25 килограммов в год составляет около 600 тысяч евро.
- И есть люди, которые его покупают?
Со слов компании "PTC", производящей препарат, его принимают до тысячи человек. В США он пока не одобрен, а в Евросоюзе получил одобрение с условием, что компания проведет еще одно клиническое испытание. В Россию компания пока не обращалась за регистрацией. Даже если они проявят желание и подадут документы, этот процесс может занять несколько лет. В России есть несколько пациентов, которым препарат "Трансларна" был закуплен при помощи благотворительных фондов. Пока же планируется клиническое исследование, куда мы постараемся включить максимально возможное количество наших пациентов.

- Какие еще испытания проводятся сейчас в мире?
Их очень много. Во французском Институте миологии сейчас ведутся испытания на животных, которых заражают вирусными векторами, несущими микродистрофин. Эту генетическую структуру сажают на аденовирус, из которого предварительно выделяют все паталогические ДНК, и затем он должен заразить каждую клеточку организма, чтобы в ней начался синтез дистрофина.
Проблема в том, что аденовирус не способен заключить в себя всю нуклеотидную последовательность гена - настолько она огромная. Поэтому может использоваться только микроген, что позволит лишь перевести форму Дюшенна в более сохранную форму Беккера, а не полностью восстановить синтез дистрофина. Но МРТ и биопсии мышц показывают, что в результате исследований у лабораторных животных некоторые мышечные клетки действительно начинают вырабатывать дистрофин, и это очень хорошо.
Есть и гипотезы, связанные со стволовыми клетками. Делая МРТ мышц, мы видим, что до пяти-шести лет у пациентов с миодистрофией Дюшенна мышцы не изменены. Возможно, это происходит за счет работы стволовых клеток. Когда мышечная клетка гибнет, на ее место приходит стволовая, пытается заместить ее и как-то работать. И так происходит, пока запас стволовых клеток не истощается.
Но пока это только гипотеза. Существует идея использования "уснувшего" гена - утрофина. Это такой эмбриональный дистрофин - он работает, только когда плод находится в эмбриональном состоянии, а затем инактивируется. Если каким-то образом снять блокировку и вновь заставить его функционировать, то он вполне может замещать неполноценный белок дистрофин и восстанавливать нормальную работу мышц.
- А какие исследования проводятся в России?
К сожалению, у нас наблюдается гигантский провал в этом научном направлении. Если государство не тратит ничего на свою науку, то потом придется тратить огромные деньги за чужую. И в 1990-е, и в 2000-е годы, и до сих пор на изучение нервно-мышечных заболеваний не выделяется никаких бюджетных денег.
Наше отделение из 30 коек целиком создано на энтузиазме сотрудников. Но мы клиницисты, мы не можем в стационаре разрабатывать препараты, работать с молекулами, лабораторными животными. Это работа для молекулярных биологов, ветеринаров, фармацевтов, провизоров-технологов и других. И только после того как будет доказана безопасность испытываемой молекулы-препарата, проводятся клинические испытания первой, второй и третьей фазы, в которых уже могут принимать участие пациенты.
- Кто же оплачивает зарубежные исследования?
Например, во Франции Институт миопатии живет только за счет пожертвований. Ежегодно в декабре родители детей с нервно-мышечными заболеваниями устраивают мощную благотворительную акцию "Телетон", в которой принимают участие звезды шоу-бизнеса, популярные телеведущие, актеры. Каждый желающий может в прямом эфире позвонить и пожертвовать любую сумму на исследования. В результате ежегодно они собирают миллионы евро!
Их примеру последовали в США, Великобритании, Италии. У нас в этом смысле больше распространена адресная помощь. Люди собирают деньги на конкретную операцию, покупку кресла для конкретного ребенка, но не на научные исследования. В России априори считается, что всю научную деятельность ведут государственные НИИ и там что-то наверняка делается, хотя это совсем не так.
Единственные российские научные исследования по Дюшенну, которые сейчас ведутся, - это малоизвестный частный проект, у которого только один спонсор - отец больного мальчика.
- Вам психологически легко работать с больными детьми, зная о том, что их ждет?
Конечно, детей очень жалко, но кто-то же должен им помогать. Когда я был молодым специалистом, то одно время думал, что схожу с ума. Было такое впечатление, что все дети вокруг больные. Тогда я устроился в поликлинику на четверть ставки, ведь там на приеме были одни здоровые дети! Легко и приятно смотреть на здоровых детей! И постепенно в голове все устаканилось.
На самом деле гораздо труднее общаться с родителями. У наших родителей первая реакция - неприятие. Они не верят диагнозу, считают его ошибочным, некоторые едут перепроверяться в Израиль или США. Думаю, это срабатывает механизм психологической защиты. А вот с теми родителями, которые принимают заболевание, работать уже значительно легче.
- Если бы у вас была возможность перебраться за рубеж, вы бы ею воспользовались?
Такая возможность была, но все-таки только здесь я ощущаю себя на своем месте. Да, существуют различные ограничения, но я делаю все, что от меня зависит. Принимаю пациентов, читаю лекции студентам на кафедре неврологии, нейрохирургии и медицинской генетики педиатрического факультета РНИМУ имени Н.И. Пирогова. И мечтаю, что когда-нибудь и в России появится нервно-мышечный центр.
Мнение эксперта
Елена Шеперд. Соучредитель фонда для детей с миодистрофией Дюшенна "МойМио"
Первая и основная программа фонда - "Мы вместе". Это, например, проведение психолого-реабилитационных лагерей. В семьях, где ребенок с редким диагнозом лишен медицинского сопровождения, встречи с коллективом высокопрофессиональных специалистов трудно переоценить. Но если для москвичей мы регулярно проводим встречи в родительском клубе, то с жителями регионов в этом смысле сложнее.
Поэтому дважды в год, осенью и весной, семьи со всей России приезжают на неделю в пансионат в Калужской области. Ребята знакомятся, рукодельничают, проходят тренинги, организуют квесты, а в других аудиториях родители общаются со специалистами. В лагерь приезжают детский и взрослый психологи, невролог, кардиолог, пульмонолог - штучные профессионалы, которых в России можно по пальцам пересчитать. Участие в программе бесплатно для всех семей, в которых есть ребенок с миодистрофией Дюшенна, фонд также берет на себя транспортные расходы.
В этом году у нас стартовала медицинская программа "Клиника МДД". Очередь в федеральное лечебное учреждение для наших ребят сейчас составляет примерно год, а для прогрессирующего заболевания - это совершенно неприемлемый срок. Теперь у нас появилась возможность за два дня провести полное обследование тех детей, которые находятся в этом списке ожидания, или подопечных ребят старше 18 лет, которым и обратиться некуда.
В Европе с миодистрофией Дюшенна живут в среднем около 30 лет. К сожалению, в России ребята часто уходят в возрасте двенадцать-четырнадцать-шестнадцать лет из-за того, что им оказывается помощь без учета особенностей заболевания. Например, после потери способности к хождению у ребят быстро слабеют диафрагмальные мышцы, снижается кашлевой рефлекс.
Ребенок с миодистрофией Дюшенна. (Канада)

В результате ребята не могут самостоятельно кашлять, чтобы откашливать мокроту при простуде или гриппе. И, если врач назначит муколитик, увеличивающий мокроту, ребенок не сможет откашлять ее и начнет задыхаться. В лучшем случае ему сделают дырку в горле и он будет дышать через трахеостому. Еще дадут кислород без контроля газов крови, чем только ухудшат ситуацию. В худшем его не спасут.
Чтобы такого не случалось, медицинскому персоналу необходимо знать особенности этого заболевания и особенности оказания помощи. Семье нужно учиться правильно жить с заболеванием. Респираторные упражнения и физическая терапия, дренажный массаж и откашливатель. В этом залог качества и продолжительности жизни ребят с МДД. Благодаря этому за рубежами нашей родины ребята живут в среднем на 10 лет дольше.
Ребятам нужны индивидуальные ортопедические коляски, регулярный прием лекарств. Все это родители вынуждены оплачивать сами, так как пока в России миодистрофия Дюшенна не имеет стандарта оказания медицинской помощи и ребята лишены системной медицинской помощи. Но сначала необходимы самостоятельные клинические рекомендации по ведению пациентов с мышечной дистрофией Дюшенна. Затем стандарт оказания медицинской помощи. На сегодняшний день это одна из самых главных задач.
Денис Решетов. Биоинженер и биоинформатик, директор по науке компании "Марлин Биотех"
Наша компания возникла три года назад, когда у сына моего знакомого диагностировали миодистрофию Дюшенна. Выяснилось, что заболевание никак не лечат, есть лишь методы поддерживающей терапии, но прогноз все равно неблагополучный.
Экзон-скиппинг 51-го экзона, который в тот момент разрабатывали Prosensa и Sarepta, не подходил нашему пациенту. Мы решили сделать это по-другому, повторяли описанные эксперименты раз за разом, но смогли убедиться только в том, что этот подход не работает, соответственно, надо искать новый.
Полтора года назад благодаря новым технологиям стало возможно доставлять в клетки генные конструкции с помощью аденоассоциированных вирусов. Сейчас мы разрабатываем препарат, который будет доставлять микроген в клетки. Пока проходят эксперименты на мышах, и, как только мы поймем, что технология работает, начнем синтезировать вирусы в больших количествах и приступим к доклиническим исследованиям.
Конечно, мы не единственная организация, которая развивает вирусный подход. Но обычно делают упор на микродистрофин, а мы больше работаем с микроутрофином. По нашим ожиданиям, этот подход подойдет всем пациентам, независимо от вида мутации.
Миодистрофия Дюшенна (МДД) - наследственное заболевание, которое начинается в возрасте 2-5 лет и характеризуется прогрессирующеймышечной слабостью, атрофией и псевдогипертрофией проксимальных мышц , нередко сопровождается кардиомиопатиями и нарушением интеллекта. На ранних этапах заболевания наблюдается повышенная утомляемость при ходьбе, изменение походки («утиная походка»). При этом происходит постепенная деградация мышечных тканей. 95% больных перестают ходить в возрасте 8-12 лет. В возрасте 18-20 лет больные, как правило, умирают, часто от дыхательной недостаточности. Выделяют аллельную МДД форму - мышечную дистрофию Беккера (МДБ, OMIM ), которая характеризуется сходными клиническими проявлениями, более поздним началом (примерно в 10-16 лет) и более мягким течением. Такие больные часто сохраняют способность ходить до 20 лет, а некоторые - до 50-60 лет, хотя в патологический процесс вовлечены те же мышцы, что и при МДД. Продолжительность жизни таких больных сокращена незначительно.
Биохимическим маркером заболевания является повышенный (в 100-200) раз уровень креатинфосфокиназы (КФК ) в крови. У носительниц поврежденного гена уровень КФК в среднем также несколько повышен.
Тип наследования мышечной дистрофии Дюшенна - Х-сцепленный рецессивный, т.е. им страдают почти исключительно мальчики, женщины же с поврежденным геном в одной из Х-хромосом являются носительницами МДД. Но в редких случаях миодистрофией Дюшенна могут болеть и девочки. Причинами этого могут быть преимущественная инактивация Х-хромосомы с нормальным аллелем у гетерозиготных носительниц мутантного гена DMD, Х-аутосомная транслокация, затрагивающая этот ген, гемизиготность по мутантному аллелю и наличие фенокопий (заболеваний, связанных с нарушением других белков, входящих в дистрофин-гликопротеиновый комплекс). Приблизительно в 2/3 случаев сын получает хромосому с повреждением от матери-носительницы, в остальных случаях заболевание возникает в результате мутации de novo в половых клетках матери или отца, либо в предшественниках этих клеток. Мышечная дистрофия Дюшена (МДД) встречается приблизительно у одного из 2500-4000 новорожденных мальчиков.
Ген DMD, ответственный за прогрессирующую мышечную дистрофию Дюшена/Беккера (МДД/МДБ), находится в локусе Хр21.2, имеет размер 2,6 млн п.н. и состоит из 79 экзонов. В 60% случаев мутации, приводящие к МДД/МДБ, представляют собой протяженные делеции (от одного до до нескольких десятков экзонов), в 30% случаев - точковые мутации и в 10% случаев - дупликации. Из-за наличия так называемых «горячих участков» делеций амплификация 27 экзонов и промоторной области гена DMD позволяет выявлять примерно 98% всех крупных делеций. Поиск точковых мутаций затруднен из-за большого размера гена и отсутствия мажорных мутаций.
В Центре Молекулярной Генетики проводится измерение уровня КФК в крови, а также прямая диагностика МДД/МДБ, представляющая собой поиск крупных делеций/lдупликаций во всех экзонах гена DMD и поиск «точковых» мутаций гена DMD методом NGS (next generation sequensing). Исследование методом NGS позволяет так же выявлять делеции всех экзонов гена DMD у больных мальчиков. Анализ всех экзонов гена позволяет определить точные экзонные границы делеции в сучае ее выявления, и таким образом, установить приводит ли данная делеция к сдвигу рамки считывания белка, что в свою очередь важно для прогноза формы заболевания - миодистрофия Дюшенна или Беккера. Таким образом, сочетание различных методов исследования позволяет выявлять практически все мутации гена DMD.
Наличие любого типа мутаций (делеции/дупликации в одном или нескольких экзонах, «точковые» мутации) является молекулярно-генетическим подтверждением клинического диагноза миодистрофии Дюшена/Беккера и позволяет проводить дородовую диагностику в данной семье.
Внимание! Для измерения уровня КФК кровь должна быть свежей (не замороженной)!
В случае дородовой диагностики необходим биоматериал плода, в качестве которого можно использовать ворсины хориона (с 8-й до 12-й недели беременности), амниотическую жидкость (с 16-й до 24-й недели беременности) или пуповинную кровь (с 22-й недели беременности).
Нами разработаны . Наборы предназначены для использования в диагностических лабораториях молекулярно-генетического профиля.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1 . Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий - около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Данная патология означает нарушение питания мышц. Указанная патология является наследственной слабостью мышц, попадается крайне редко.
Этот синдром обычно мучает лишь мальчиков. 1 заболевание приходится на 3000 случаев рождения нормальных малышей.
Имеются другие разновидности мышечных дистрофий, которые возникают у девочек, но там проявления легче.
В ходе данного заболевания, при прогрессировании его возникает нарушение связей между мышцами и нервами.
Синдром Дюшена передаётся по наследству, мать - является носителем гена описываемой патологии, но не болеет сама.
Другие виды дистрофии
Кроме описываемого синдрома, имеются и другие типы данных дистрофий, хотя они редко возникают.
Синдром Беккера - крайне редок, и так же болеют лишь мужские особи. Патология возникает в 10-11 лет, и становится менее заметной в 35-40 лет.
Наследственная мускульная миопатия - заболевают и девочки и мальчики, так же имеет генетическую причину, наблюдается даже реже, чем описываемый синдром.
Плече-лопаточно-лицевая миопатия - крайне долгое развитие, течение благоприятное. Появляется до 10 лет. Характеризуется: будучи в грудном возрасте не могут хорошо высосать грудь; в более позднем возрасте не получается сделать губы трубочкой; тяжело осуществлять подъем рук, лицо имеет маскообразный вид.
Дистрофия Эмери—Дрейфуса - проявляется, как и остальные типы дистрофий, но в отличие от предыдущих, эти плохо влияют на сердце.
Первые симптомы
Ребёнок позже ходит, и то, все его попытки оказываются не успешными. Ребёнок ходит, переваливаясь из одной стороны в другую, часто падает на попу, желание подняться и встать из положения тела, сидя на полу, зачастую оказываются неудачными.
Мускулы ног могут выглядеть довольно крепкими, хотя в реальности таковыми не являются. Все остальные мышцы, отвечающие за ходьбу - развиты плохо.
Диагностика
Видя характерные симптомы, врач может подозревать у малыша данную патологию. В таком случае терапевт должен дать направление ребенку к ортопеду.
Исследование крови: в нормальной мышечной материи имеется креатин фосфокиназа. При данной патологии количество этого фермента слишком завышено.
Мышечный тест: при помощи электронного исследования мышц меряется быстрота передачи нервных реакций в мускулы.
Биопсия мышц: кусочек мышечной ткани рассматривается под микроскопом. При таком исследовании выявляются наросты в виде атеросклеротических бляшек.
Почему эта патология считается генетической
Указанный синдром, генетическое расстройство, появляющиеся из-за видоизменённой аллели в половой Х-хромосоме.
Любая клеточка человеческого организма, за исключением гамет содержит 46 хромосом. Одна аутосома содержит в своём составе множество алелий (около 1000). В аллелях и аутосомах расположена дезоксирибонуклеиновая кислота, ответственная за передачу данных, переходящих из одного поколения в другое.
Структура гена белковая. Глобулины — строительный материал для человеческого организма.
Данная болезнь появляется из-за нахождения специфического гена в Х-хромосоме, которая находится как в мужском, так и в женском организме. Аллель реагирует на продуцирование белка, формирующего нормальную мускульную ткань.
Девочки иногда получают в наследство изменённый аллель, но обычно, болезнь Дюшена у них не появляется, так как в их организме имеются две Х-хромосомы, одна из них, здоровая, замещает нарушение во второй.
В ходе определённых экспериментов были получены следующие данные, болезнь Дюшена у мальчиков бывает врожденной, и приобретенной из-за мутаций, случившихся в генотипе после рождения.
Прогрессирование патологии
С прогрессированием патологии, признаки выражаются более отчётливо, это происходит потому, что мышцы уже практически не могут способствовать адекватному перемещению. Со временем мышцы рук ослабевают, и малышу очень трудно становится брать и держать предметы. Мускулы рук и ног становятся дистрофичными, суставы приобретают тугоподвижность. Не редко возникает деформация локтевых, тазобедренных и коленных суставов. Мускулы, удерживающие позвоночный столб, перестают расти, из-за чего позвоночник приобретает изгибы. Малышу сложно ходить. Кой-какие дети плохо учатся. Научение вызывает кропотливый труд, в ходе чего ребёнок просто не успевает за учебной программой.
Как помочь малышу?
К сожалению, этот синдром не лечиться, поэтому стоит подсказать ребёнку, как адаптироваться к жизни с данной болезнью.
Ребёнку показана психотерапия, дабы убедить его в важности занятий физической нагрузкой.
Стоит упражняться в занятиях специальной гимнастикой, дабы сохранить движения в суставах.
Чтобы не образовывались контрактуры, стоит применять специальные шины и корсеты.
Показана лечебная ходьба.
Дети с указанным синдромом, испытывают некоторые трудности в научении, поэтому им необходим индивидуальный подход и внимание. Хорошо, если больной малыш станет учиться в специальной школе, для детей с данными трудностями. Существуют такие школы -интернаты, программа в которых адаптирована именно для такого контингента.
Если у больного малыша имеются братья или сёстры, им необходимо в равной степени уделять внимание, чтоб дети не думали, что они брошены.